.png) Agenzia Italiana del Farmaco
Agenzia Italiana del Farmaco
Ispezioni di Farmacovigilanza
Attraverso le ispezioni di Farmacovigilanza (Good Pharmacovigilance Practices, GVP) l’AIFA verifica la conformità delle aziende farmaceutiche alle disposizioni della normativa italiana e comunitaria in materia di farmacovigilanza, secondo quanto previsto dal Decreto ministeriale 30 aprile 2015. Le ispezioni possono riguardare i locali, le registrazioni, i documenti e il fascicolo di riferimento del sistema di farmacovigilanza del titolare dell'Autorizzazione all’Immissione in Commercio o di terzi incaricati dallo stesso titolare di realizzare le attività a cui si fa riferimento nel decreto su indicato.
Un altro compito degli ispettori GVP è quello di condurre ispezioni su studi clinici con problematiche correlate alla farmacovigilanza, con particolare attenzione agli Studi PASS (Post-authorisation safety study).
Grande importanza riveste la formazione professionale degli ispettori, anche attraverso la partecipazione a corsi promossi dall’EMA.
Le tipologie di ispezione GVP
Ci sono tre tipi di ispezioni GVP alle aziende farmaceutiche :
- Ispezioni di routine nazionali: sono ispezioni eseguite presso i titolari dell'autorizzazione all'immissione in commercio in Italia e incluse nel programma annuale dell’AIFA. Il titolare riceve la notifica di tali ispezioni in anticipo. Gli ispettori verificano i sistemi e le procedure utilizzate da un titolare per adeguarsi alle vigenti normative e alle linee guida in materia di Farmacovigilanza.
- Ispezioni straordinarie (Triggered) nazionali: sono ispezioni, che vengono eseguite, per esempio, dopo segnalazioni circa problemi di sicurezza oppure per sospette violazioni della normativa in materia di monitoraggio della sicurezza dei medicinali o su richiesta da parte di altri uffici dell’AIFA. In rari casi, la notifica di tali ispezioni ai titolari dell'autorizzazione all'immissione in commercio potrebbe non essere inviata in anticipo.
- Ispezioni richieste dal Committee for Medicinal Products for Human Use (CHMP) dell’EMA per medicinali autorizzati con procedura centralizzata. Queste ispezioni possono essere di routine o straordinarie (Triggered). L'organizzazione generale e il processo per le ispezioni di farmacovigilanza richieste dal CHMP sono descritte nella Union procedure on the coordination of EU pharmacovigilance inspections (EMA/INS/PhV/192234/2014) pubblicata sul sito web dell’EMA.
Documentazione richiesta ai titolari AIC in fase pre-ispettiva
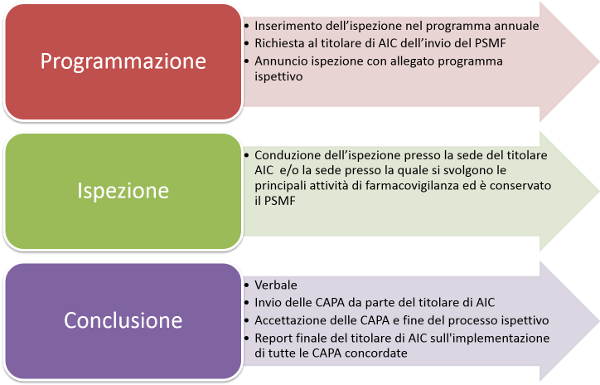
I titolari dell'AIC sono tenuti a compilare e presentare all’AIFA il Fascicolo di riferimento del Sistema di Farmacovigilanza (PSMF) prima dell'ispezione. Il PSMF fornisce informazioni per aiutare sia il titolare sia gli ispettori per la preparazione dell'ispezione. Per una corretta compilazione del PSMF, si invita a far riferimento alla relativa linea guida (GVP Module II – Pharmacovigilance system master file) pubblicata sul sito web dell’EMA.
Le ispezioni sono costituite da visite in loco durante le quali vengono eseguiti colloqui, esaminati documenti e raccolte informazioni sui sistemi informatici utilizzati per la Farmacovigilanza.
Il verbale di ispezione GVP
Dopo l’ispezione viene preparato un verbale che viene inviato al titolare di AIC interessato e nel quale le eventuali deviazioni sono classificati in critiche, maggiori e minori, in linea con quanto concordato in ambito Unione Europea:
- Critiche: deficienze nei sistemi, pratiche o processi di farmacovigilanza che influiscono negativamente sui diritti, la sicurezza, la salute e il benessere dei pazienti o che comportino un rischio potenziale per la salute pubblica. Grave inottemperanza a rilevanti requisiti normativi e alle linee guida.
- Maggiori: deficienze nei sistemi, pratiche o processi di farmacovigilanza che potrebbero influire negativamente sui diritti, la sicurezza, la salute e il benessere dei pazienti o che potrebbero comportare un rischio potenziale per la salute pubblica. Inottemperanza a rilevanti requisiti normativi e alle linee guida.
- Minori : deficienze nei sistemi, pratiche o processi di farmacovigilanza che non dovrebbero influire negativamente sui diritti, la sicurezza, la salute e il benessere dei pazienti o che non dovrebbero comportare un rischio potenziale per la salute pubblica.
Quali sono i compiti dell’ispettorato GVP dell’AIFA?
I compiti dell’Ispettorato GVP dell’AIFA, istituito il 1 Aprile 2010 per ottemperare a quanto previsto dal D.L.vo 24 aprile 2006 n°219 all’art.134, come da Decreto del Ministero della salute del 30 aprile 2015, art 42, sono:
- condurre ispezioni al fine di valutare la conformità delle aziende farmaceutiche con la normativa italiana e la legislazione comunitaria in materia di monitoraggio della sicurezza dei medicinali somministrati ai pazienti;
- le ispezioni possono riguardare i locali, le registrazioni, i documenti, il fascicolo di riferimento del sistema di farmacovigilanza (PSMF) del titolare AIC o di terzi incaricati dallo stesso titolare di realizzare le suddette attività;
- dopo ogni ispezione redigere una relazione ispettiva in cui gli ispettori riportano se il titolare AIC o i terzi incaricati dallo stesso titolare rispettano le disposizioni normative vigenti;
- condurre ispezioni su studi clinici osservazionali (PASS - Post Authorization Safety Studies);
- cooperare con l’EMA nello scambio di informazioni sia sulle ispezioni programmate che sulle ispezioni condotte;
cooperare con le autorità competenti degli altri Stati membri e con l’EMA nel coordinamento delle ispezioni nei Paesi terzi.
Quali sono le tipologie di ispezione condotte dall’Ispettorato GVP?
Il programma nazionale delle ispezioni include sia le ispezioni di routine che fanno parte del programma europeo, sia le ispezioni ai titolari dell'AIC che detengono solo autorizzazioni nazionali. Inoltre comprende ispezioni straordinarie a seguito di segnalazioni provenienti da fonti diverse (Autorità Giudiziarie, altri Uffici dell’AIFA o di altre strutture pubbliche, CHMP dell’EMA e/o Commissione Europea).
Le tipologie di ispezioni condotte dall’Ispettorato GVP dell’AIFA sono:
- ispezioni di sistema
- ispezioni prodotto specifiche
- ispezioni di routine
- ispezioni pre-autorizzazione
- ispezioni post-autorizzazione
- ispezioni annunciate
- ispezioni non annunciate
- ispezioni “for cause”
- re-ispezioni
ispezioni a distanza (remote inspections).
Quando si ritiene conclusa un’ispezione?
Il processo ispettivo si ritiene concluso con l’approvazione da parte del team ispettivo del piano di azione preventive e correttive delle deficienze riscontrate (CAPA plan) predisposto dall’azienda ispezionata. L’azienda è tenuta comunque ad inviare un report finale alla conclusione di tutte le azione correttive all’Ufficio Ispezioni GVP.
Quali sono i requisiti, i compiti e le responsabilità della persona responsabile della farmacovigilanza (QPPV)?
Come previsto dalla Direttiva 2010/84/EU, dal Regolamento UE n. 520/2012, dall’art.17 del Decreto 30 aprile 2015, nell’ambito del sistema di farmacovigilanza, il titolare dell’autorizzazione all’immissione in commercio deve disporre a titolo stabile e continuativo di una persona responsabile della farmacovigilanza (QPPV):
- adeguatamente qualificata dal punto di vista teorico e pratico per lo svolgimento delle attività di farmacovigilanza;
- con documentata esperienza in tutti gli aspetti di farmacovigilanza;
- i compiti della QPPV devono essere definiti in un mansionario e i rapporti gerarchici con il personale direttivo e di controllo definiti in un organigramma;
- le informazioni relative alla QPPV devono essere riportate nel fascicolo di riferimento del sistema di farmacovigilanza (PSMF);
- se la persona qualificata per la farmacovigilanza non ha compiuto la formazione medica di base, il titolare dell’AIC si assicura che sia assistita da una persona in possesso di formazione medica e tale assistenza è debitamente documentata;
- la persona qualificata per la farmacovigilanza risiede e svolge la propria attività nell’Unione europea;
- la persona qualificata per la farmacovigilanza è responsabile dell’istituzione e della gestione del sistema di farmacovigilanza;
- il titolare dell’autorizzazione comunica il nominativo e le informazioni per contattare la persona qualificata all’AIFA e all’EMA;
- la persona qualificata per la farmacovigilanza riceve una formazione iniziale e continua in relazione al suo ruolo e alle sue responsabilità;
La linea guida GVP Modulo I, paragrafi C.1.1, C.1.2 e C.1.3 dettaglia le responsabilità, le qualifiche e il ruolo della persona qualificata per la farmacovigilanza (QPPV).
Il titolare AIC, tramite il database dell’art. 57, notifica all’EMA le modifiche relative alla QPPV.
Qual è la responsabilità del titolare dell'autorizzazione all'immissione in commercio in relazione alla persona qualificata responsabile della farmacovigilanza?
Il titolare dell’autorizzazione all’immissione in commercio si assicura che la persona qualificata responsabile della farmacovigilanza abbia l’autorità sufficiente per influire sul funzionamento del sistema di qualità e delle attività di farmacovigilanza.
Che cosa si deve prevedere in caso di assenza della QPPV?
Il Regolamento n. 520/2012 all’art.2, comma 1 lettera d) prevede che nel PSMF sia descritta la procedura da seguire in caso di assenza della QPPV e la GVP Modulo I, paragrafo C.1.3 specifica che la persona che sostituisce la QPPV ( la cosiddetta Deputy QPPV) ,in caso di sua assenza, deve essere indicata nel fascicolo di riferimento del sistema di farmacovigilanza (PSMF) unitamente ai suoi riferimenti e deve avere conoscenza di tutte le informazioni necessarie a ricoprire tale ruolo.
Che cosa è il fascicolo di riferimento del sistema di farmacovigilanza (PSMF)?
Il fascicolo di riferimento del sistema di farmacovigilanza (PSMF) è il documento che descrive dettagliatamente il Sistema di farmacovigilanza utilizzato dal titolare dell’autorizzazione all’immissione in commercio per uno o più medicinali autorizzati a norma della Direttiva 2001/83/EC o del Regolamento CE n. 726/2004. I contenuti del PSMF sono descritti nel dettaglio nella linea guida GVP Modulo II.
Dove deve essere conservato il fascicolo di riferimento del sistema di farmacovigilanza (PSMF)?
Come da Regolamento UE n. 520/2012 il PSMF deve essere conservato nel luogo dell’Unione in cui sono svolte le principali attività di farmacovigilanza del titolare dell’autorizzazione all’immissione in commercio o nel luogo dell’Unione in cui opera la persona qualificata per la farmacovigilanza (QPPV).
Il titolare dell’autorizzazione all’immissione in commercio ha l’obbligo di tenere aggiornato e mettere a disposizione, su richiesta un fascicolo di riferimento di farmacovigilanza per uno o più medicinali di cui all’art. 17 del decreto 30 aprile 2015.
Il titolare AIC, tramite il database dell’art. 57, notifica all’EMA le modifiche relative al luogo in cui è conservato il PSMF.
Quando è necessario aggiornare il database art. 57? Con quali tempistiche?
Secondo la GVP Modulo II (II.B.2.2. Location, registration and maintenance) il titolare AIC deve assicurare che le informazioni riguardanti il PSMF e QPPV inserite nel database art.57 siano costantemente aggiornate.
In caso di variazione della QPPV o delle informazioni riguardo al PSMF location, il titolare deve aggiornare tali informazioni all’interno del database immediatamente o comunque entro 30 giorni di calendario dalla variazione.
Per quanto riguarda invece le informazioni relative ai medicinali, è necessario aggiornare il database art. 57 entro 15 giorni per le nuove AIC ed entro 30 giorni per modifiche alle AIC esistenti (variazioni, trasferimento di titolarità, rinnovi, sospensioni, revoche e ritiro) come indicato dal “Legal notice on the implementation of Article 57(2) of Regulation (EC) No. 726/2004” (EMA/505633/2011, Rev. 2) disponibile al seguente indirizzo: https://www.ema.europa.eu/documents/other/legal-notice-implementation-article-572-second-subparagraph-regulation-ec-no-726/2004_en.pdf (ultimo accesso verificato 29/11/2018).
Quali sono i riferimenti normativi relativi all’archiviazione dei documenti di farmacovigilanza?
Come definito dal regolamento 520/2012 art. 12, i titolari di AIC sono tenuti alla registrazione di tutti i documenti utilizzati per le attività di farmacovigilanza e alla loro custodia. A tal proposito i titolari di AIC predispongono un sistema di gestione (Archivio) che permetta di reperire tale documentazione e di rintracciare come sono state affrontate le problematiche di farmacovigilanza, nonché le decisioni prese in merito. I dati di farmacovigilanza e i documenti relativi ai singoli medicinali autorizzati sono conservati fintanto che il prodotto è autorizzato e almeno per i dieci anni seguenti la scadenza dell’autorizzazione all’immissione in commercio. Tuttavia, i documenti sono conservati per un periodo più lungo se la legislazione dell’Unione o la legislazione nazionale lo richiedono.
Da quando è in vigore il repository EMA per l’invio degli PSUR?
A partire dal 13 giugno 2016 è disponibile il repository per l’invio degli PSUR predisposto da EMA utilizzando l’eSubmission Gateway Web Client. Il caricamento degli PSUR nel repository rappresenta l’unica modalità con cui devono essere presentati gli PSUR dei medicinali autorizzati in UE, a prescindere dalla procedura autorizzativa e inclusi i medicinali autorizzati solo in uno Stato membro dell’UE.
Quali sono le tempistiche entro cui presentare uno PSUR?
Il titolare AIC è tenuto a presentare uno PSUR a scadenze definite a partire dalla data di autorizzazione. In accordo a quanto previsto dalla Direttiva 2010/84/EU l’EMA gestisce e pubblica la lista (EU Reference Dates – EURDlist) delle date e frequenze di presentazione degli PSUR per i principi attivi contenuti nei medicinali autorizzati nell’Unione Europea in almeno due Stati membri. Per i principi attivi non elencati nella EURD list (o nella Extended list), autorizzati in un solo Stato Membro, la presentazione dello PSUR è soggetta alla frequenza di invio standard prevista dalla normativa vigente (art. 107c della Direttiva 2010/84/EU) come specificato al capitolo VII.C.2. “Standard submission schedule of PSURs” del Modulo VII delle GVP - Periodic safety update report, salvo condizioni diverse imposte all’atto della autorizzazione all’immissione in commercio (e/o in sede di rinnovo o in seguito alla valutazione di PSUR/PBRER nell’ambito della procedura PSUSA) dall’autorità competente.
L’EURD list, in formato excel, è disponibile alla seguente pagina dedicata agli PSUR. Gli aggiornamenti dell'EURD List sono adottati dopo consultazione del PRAC.
Dove posso reperire le informazioni riguardo gli outcome delle procedure PSUSA?
Tutti gli esiti delle procedure di valutazione degli PSUR (PSUR Single Assessment, PSUSA) sono pubblicamente disponibili sul sito dell’EMA.
In particolare, nella sezione “Periodic safety update report single assessments” è disponibile, in formato excel, l’elenco delle procedure PSUSA per tutti i principi attivi contenuti nei medicinali autorizzati tramite procedura di autorizzazione nazionale. In alternativa al formato tabellare è possibile reperire la procedura PSUSA dei principi attivi contenuti nei medicinali autorizzati tramite procedura nazionale con motore di ricerca disponibile al seguente link.
Dove è possibile reperire le informazioni sulla ricerca di letteratura effettuata da EMA?
Dal 1 Settembre 2015 l'EMA è responsabile, ai sensi dell'Art. 27 del Regolamento EU 726/2004, del monitoraggio di una serie di riviste medico-scientifiche internazionali al fine di identificare reazioni avverse correlate a principi attivi contenuti in medicinali autorizzati nell'Unione Europea e inserire tali dati in Eudravigilance. La lista esaustiva di sostanze attive e la selezione della letteratura monitorata da EMA sono disponibili al seguente link. Seguendo le modalità descritte nel modulo VI delle GVP, i titolari di AIC sono responsabili del monitoraggio della letteratura scientifica relativamente ai principi attivi non compresi nella suddetta lista di EMA nonché del monitoraggio, per i principi attivi compresi nella lista di EMA, della letteratura di carattere locale.
Dove è possibile reperire la lista IME (Important Medical Event)?
La lista IME, che fornisce un supporto per la classificazione e la definizione della gravità di una sospetta reazione avversa ed è inoltre basata sui termini medici adottati da MedDRA (Medical Dictionary for Regulatory Activities), è disponibile al seguente link.
Dove è possibile reperire le raccomandazioni del PRAC sui segnali?
L’EMA pubblica mensilmente un resoconto riepilogativo dei segnali di sicurezza discussi durante l’ultima riunione del PRAC, e le raccomandazioni emesse per ciascun segnale. Le raccomandazioni del PRAC talvolta comportano un’azione regolatoria a carico del/i medicinale/i contenenti i principi attivi coinvolti (es. modifiche delle informazioni del prodotto nel Riassunto delle Caratteristiche del Prodotto e nel Foglio Illustrativo) che deve essere implementata dai titolari AIC mediante apposita variazione. Al seguente indirizzo sono pubblicati gli estratti della raccomandazione del PRAC comprensiva dei testi della modifica delle informazioni del RCP e FI in lingua inglese e nelle traduzioni in lingua nazionale. I titolari di AIC sono tenuti a verificare l’eventuale presenza di richieste di adempimenti per i prodotti di propria competenza contenenti i principi attivi oggetto di raccomandazioni e ad ottemperare alla richiesta entro le tempistiche indicate.






