.png) Agenzia Italiana del Farmaco
Agenzia Italiana del Farmaco
Procedura di autorizzazione centralizzata
La procedura centralizzata ai sensi del Regolamento (CE) 726/2004 è coordinata dall’Agenzia europea per i Medicinali (EMA), che lavora in rete con le autorità competenti di ciascuno Stato membro. L’autorizzazione così ottenuta è valida in tutti i paesi dell’UE e nei tre Stati dell’Associazione europea di libero scambio (European Free Trade Association, EFTA) dello Spazio Economico Europeo (SEE): Islanda, Liechtenstein e Norvegia.
Attraverso il suo Comitato scientifico per i Medicinali per Uso Umano (Committee for Human Medicinal Products, CHMP), l’EMA valuta la documentazione presentata dall'azienda farmaceutica, verifica il rapporto beneficio/rischio sulla base dei dati di qualità, efficacia e sicurezza del medicinale ed esprime un parere entro un arco di tempo predefinito (massimo 210 giorni). Il CHMP è composto da rappresentanti di ciascuno Stato membro e da esperti selezionati sulla base di specifiche competenze scientifiche.
L'iter di valutazione prevede il coinvolgimento attivo di due Stati membri che svolgono il ruolo di (Co-)Rapporteur in modo indipendente tra di essi. Gli altri Stati membri possono esprimere commenti alla valutazione dei (Co-)Rapporteur e l'azienda farmaceutica ha la possibilità di rispondere alle richieste di chiarimento emerse dalla valutazione collegiale del CHMP.
Il parere espresso dal CHMP, a maggioranza o all'unanimità, viene trasmesso alla Commissione europea, che emana una decisione finale sull’autorizzazione all’immissione in commercio (AIC) del medicinale con carattere vincolante per tutti gli Stati membri.
Autorizzazione subordinata a condizioni
L’autorizzazione subordinata a condizioni viene rilasciata pur non essendo disponibili dati clinici completi in merito alla sicurezza e all’efficacia del medicinale ed è subordinata a specifiche condizioni (obblighi post-autorizzativi per l'azienda farmaceutica).
Tale autorizzazione si può applicare a medicinali:
- da utilizzare in situazioni di emergenza in risposta a minacce per la salute pubblica;
- designati orfani;
- impiegati per il trattamento di malattie gravemente debilitanti o potenzialmente letali.
L'azienda farmaceutica dovrà presentare ulteriori dati in un arco di tempo predefinito in modo da confermare la positività del rapporto beneficio/rischio del medicinale basata sui dati preliminari già disponibili. L’autorizzazione subordinata a condizioni ha validità annuale e il suo rinnovo dipende dall'esito della valutazione degli ulteriori dati forniti a completamento del profilo di efficacia e sicurezza del medicinale. Potrà essere convertita in approvazione normale quando l’azienda farmaceutica avrà completato gli obblighi imposti dal CHMP.
Autorizzazione in circostanze eccezionali
Nel caso in cui fattori quali la rarità della patologia, il grado di sviluppo delle conoscenze scientifiche e i principi deontologici non consentano di raccogliere informazioni complete sull'efficacia e la sicurezza del medicinale nelle normali condizioni d'impiego, può essere rilasciata l’autorizzazione in circostanze eccezionali.
I medicinali autorizzati in circostanze eccezionali sono soggetti a specifiche misure di minimizzazione del rischio definite dal CHMP e dal Comitato per la Valutazione dei Rischi per la Farmacovigilanza (Pharmacovigilance Risk Assessment Committee, PRAC) che permettono l’individuazione rapida e puntuale di qualsiasi evento avverso/incidente connesso alla somministrazione del farmaco. Sebbene il rapporto beneficio/rischio del medicinale sia positivo, il livello di evidenza scientifica rimarrà verosimilmente preliminare e soggetto a rivalutazione annuale. Pertanto l'autorizzazione in circostanze eccezionali può non essere mai convertita in approvazione normale.
Le Relazioni di Valutazione Pubblica Europea (European Public Assessment Report, EPAR) in lingua inglese dei medicinali approvati con procedura centralizzata sono accessibili tramite il sito web dell'EMA. Oltre alle EPAR relative alla valutazione scientifica che ha portato al rilascio della iniziale (nuova) AIC, sono disponibili anche le EPAR delle variazioni dell'AIC considerate "maggiori" (es. estensione delle indicazioni terapeutiche).
Revisione ciclica
La revisione ciclica (rolling review) è una procedura regolatoria ad hoc messa in atto dall'EMA per assicurare l’accesso precoce a farmaci promettenti in contesti emergenziali. La revisione ciclica permette al CHMP di valutare i dati in modo progressivo a mano a mano che questi vengono resi disponibili dalle aziende farmaceutiche durante lo sviluppo del medicinale.
Questa modalità ha lo scopo di velocizzare la successiva fase di valutazione della richiesta di autorizzazione all’immissione in commercio o della estensione della indicazione terapeutica in caso di medicinale già autorizzato. Diversamente nel contesto normale la valutazione della richiesta di autorizzazione all’immissione in commercio o di estensione della estensione della indicazione terapeutica dei farmaci è avviata solo al momento in cui tutto l’insieme delle evidenze scientifiche è presentato all’Autorità regolatoria.
Pertanto, la revisione ciclica assicura lo stesso standard di qualità e la stessa solidità delle evidenze di efficacia e sicurezza che abitualmente sono generate a supporto delle decisioni regolatorie, ma consente una valutazione in tempi più ravvicinati.
L’applicazione di questa procedura valutativa durante la pandemia da SARS-CoV-2, ha permesso la approvazione di vaccini e medicinali anti-COVID-19 in tempi nettamente più brevi rispetto alla normale procedura di autorizzazione all’immissione in commercio dei medicinali.
Come si articola il processo di valutazione centralizzata
Dalla validazione (Giorno 1) del dossier registrativo presentato dal richiedente l'AIC (azienda farmaceutica), la valutazione scientifica fino al parere espresso dal CHMP dura 210 giorni ed è articolata come descritto di seguito:
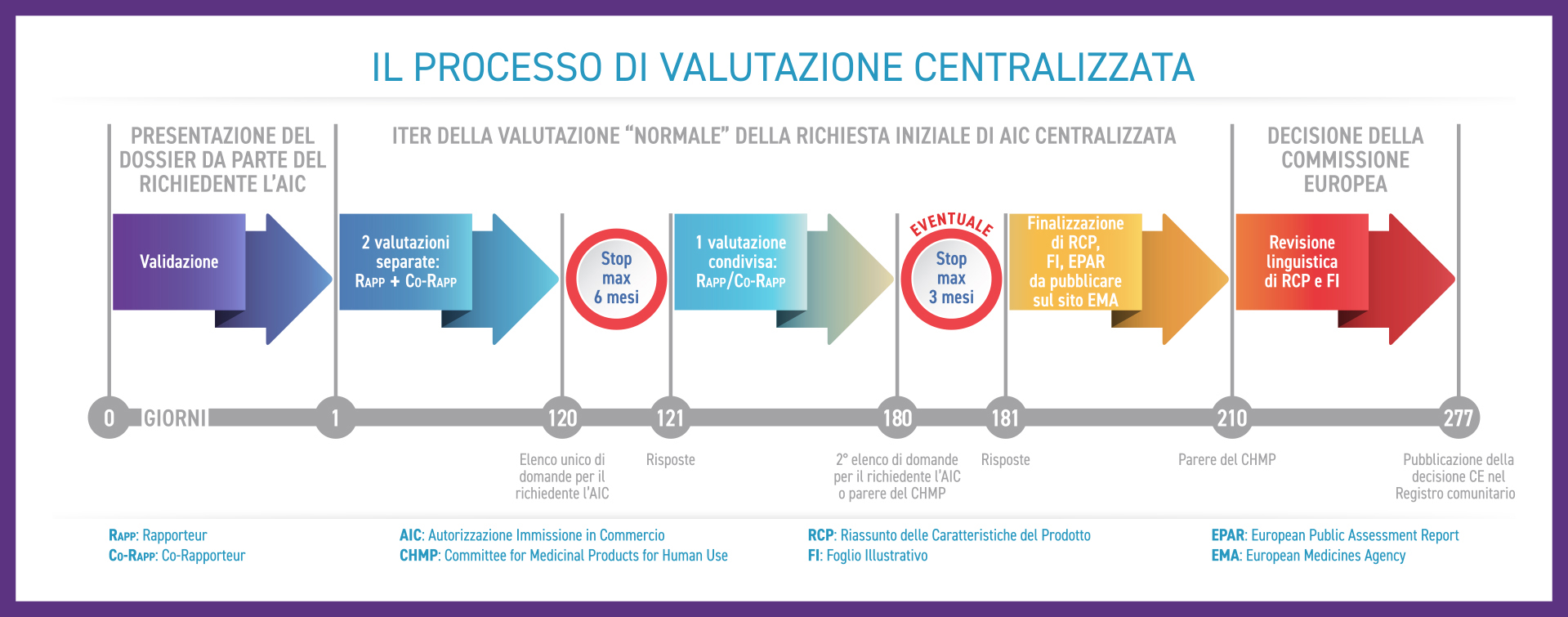
Questo tipo di procedura è definita "normale". Per medicinali di elevato interesse per la salute pubblica, in particolare sotto il profilo dell'innovazione terapeutica, per i quali esiste un bisogno clinico non soddisfatto e non sono disponibili alternative terapeutiche, può essere applicata, su motivata richiesta dell’azienda farmaceutica, la procedura di valutazione accelerata in cui i tempi previsti per il parere del CHMP si riducono a 150 giorni (il Giorno 120 della procedura normale diventa il Giorno 90).
Quando è obbligatoria la procedura centralizzata
La procedura centralizzata è obbligatoria per le seguenti classi di medicinali:
- derivati da procedimenti biotecnologici (anticorpi monoclonali, ormoni polipeptidici, emoderivati ricombinanti);
- terapie avanzate (genica, cellulare somatica, di ingegnerizzazione tissutale);
- designati orfani (farmaci utilizzati per patologie rare);
- contenenti nuove sostanze attive per il trattamento di specifiche patologie quali sindrome da immunodeficienza acquisita, cancro, malattie neurodegenerative, diabete, patologie autoimmuni, altre disfunzioni immunitarie e malattie di origine virale.
Quando la procedura centralizzata è facoltativa
La procedura centralizzata è facoltativa per le seguenti classi di medicinali:
- contenenti nuove sostanze attive per il trattamento di patologie diverse da quelle sopra menzionate;
- contenenti sostanze attive note che siano giudicati innovativi sul piano terapeutico, scientifico o tecnologico (es. nuova indicazione terapeutica, nuova forma farmaceutica);
- generici o ibridi (equivalenti e biosimilari non biotecnologici) di medicinali centralizzati già autorizzati;
- di interesse a livello comunitario (es. generici di un medicinale autorizzato a livello nazionale, vaccini pandemici).
Cosa succede dopo il parere positivo del CHMP
Al parere positivo del CHMP relativo a nuovi medicinali e a medicinali già approvati che hanno subito delle modifiche dell'AIC, segue la decisione da parte della Commissione europea che viene emessa dopo circa 60 giorni con la notifica all’azienda farmaceutica. La decisione viene pubblicata nel “Registro Comunitario dei medicinali per uso umano” accessibile dal sito web della Commissione europea, in cui vengono riportate le informazioni sul medicinale tradotte in tutte le lingue dell’UE; successivamente, ne viene data informativa nella Gazzetta Ufficiale dell’UE.
Nel caso di modifiche dell'AIC cosiddette “minori”, il parere del CHMP è direttamente applicabile e la Commissione europea emette la relativa decisione entro un anno.
Nella fase successiva al parere del CHMP, le agenzie nazionali, su richiesta dell’EMA, sono coinvolte nella revisione linguistica dei testi delle informazioni sul medicinale (riassunto delle caratteristiche del prodotto, foglio illustrativo ed etichettatura) approvati dal CHMP. Tale attività ha lo scopo di verificare la correttezza della traduzione dei testi presentati dalle aziende farmaceutiche prendendo come versione di riferimento quella in lingua inglese, e di assicurare che le informazioni tecnico-scientifiche trasferite nel foglio illustrativo siano facilmente comprensibili (leggibili) così da permettere l'uso sicuro del medicinale.
La commercializzazione in Italia
Entro 60 giorni dalla data di pubblicazione della sintesi della Decisione della Commissione europea nella Gazzetta Ufficiale dell'UE di nuove AIC, l’AIFA emette il provvedimento di classificazione nella sezione dedicata ai medicinali non ancora valutati ai fini della rimborsabilità a carico del Servizio Sanitario Nazionale (classe C non negoziata, C(nn) (art. 12, comma 5 della Legge 189/2012). Il medicinale classificato C(nn) può essere commercializzato in Italia, ma la spesa per l'acquisto non è rimborsata dal Servizio Sanitario Nazionale.
Parallelamente e indipendentemente dalla classificazione C(nn), il titolare dell'AIC centralizzata può presentare all'AIFA la domanda di rimborsabilità e di contrattazione del prezzo.
L’AIFA rende disponibile l'elenco dei farmaci valutati per l'inserimento in classe C(nn), l'elenco mensile dei medicinali in discussione alla Commissione Tecnico Scientifica e la modulistica collegata.
Per questioni inerenti la classificazione C(nn), gli interessati possono comunicare con l’Ufficio Procedure Centralizzate o il Settore Innovazione e Strategia del Farmaco utilizzando il seguente indirizzo mail: legge189.uae@aifa.gov.it o il seguente indirizzo PEC: protocollo@pec.aifa.gov.it.
Procedimenti che riguardano le informazioni nazionali
Tra le azioni di recepimento a livello nazionale dei medicinali autorizzati con procedura centralizzata, vi sono i procedimenti che riguardano le informazioni nazionali sui medicinali:
- requisiti nazionali da inserire nell'etichetta esterna (cosiddetti requisiti di Blue-Box);
- valutazione tecnico-scientifica delle informazioni addizionali richieste dai titolari dell'AIC, accessibili tramite Quick Response (QR) code posto sull'etichetta e/o foglio illustrativo ai sensi dell'art. 62 della Direttiva 2001/83/CE e s.m.i. recepito dall'art. 79 del D.Lvo. 219/2006 e s.m.i.;
- valutazione tecnico-scientifica della richiesta di esenzione dalla traduzione in lingua italiana e tedesca (per le confezioni commercializzate nella provincia di Bolzano) dell'etichetta e/o foglio illustrativo ai sensi dell'art. 63(3) della Direttiva 2001/83/CE e s.m.i. recepito dall’art. 80 comma 4 del D.Lvo 219/2006 e s.m.i.;
- notifiche all’AIFA da parte dei titolari dell'AIC di nomina dei concessionari di vendita ai sensi dell’art. 73, comma 2 del D.lvo 219/2006 e s.m.i.
Revisione scientifica ai sensi dell’art. 5.3 del Regolamento (CE) 726/2004
Su richiesta del direttore dell'EMA, della Commissione europea o di uno Stato membro, il CHMP fornisce un parere scientifico ai sensi dell'art. 5.3 del Regolamento (CE) 726/2004 su una qualsiasi questione scientifica correlata alla valutazione di un medicinale o una classe di medicinali indipendentemente dalla loro formale autorizzazione all'immissione in commercio da parte della Commissione europea. Il parere scientifico formulato sulla base dei dati disponibili di qualità, non-clinici e clinici è da intendersi di supporto alle decisioni dei singoli Stati membri.
La valutazione scientifica e le condizioni d'uso dei medicinali definite dal CHMP, sono pubblicate sul sito web dell'EMA.
Attraverso questo tipo di revisione, nel 2020 sono state definite al livello europeo le misure di minimizzazione del rischio di contaminazione dei medicinali da parte di sostanze appartenenti alla classe delle nitrosammine, e durante la pandemia da SARS-CoV-2, a partire dal 2020, sono stati emessi pareri scientifici sull’uso di medicinali non ancora approvati dalla Commissione europea o approvati in altre indicazioni terapeutiche, per la prevenzione e/o il trattamento di COVID-19.
Distribuzione temporanea di medicinali in Italia
Al fine di fronteggiare tempestivamente situazioni di emergenza sanitaria, il Ministero della salute Italiano può autorizzare la temporanea distribuzione di un medicinale per cui non è autorizzata l'immissione in commercio, ai sensi dell’art. 5.2 del Decreto Legislativo n. 219 del 24 aprile 2006.
Attraverso questo strumento regolatorio, durante la pandemia da SARS-CoV-2 è stato possibile, nelle more del perfezionamento delle procedure di autorizzazione all'immissione in commercio, rendere disponibili in Italia medicinali per la prevenzione e/o il trattamento di COVID-19. Il parere scientifico formulato dal CHMP ai sensi della revisione secondo l’art. 5.3 del Regolamento (CE) 726/2004, ha fornito la base per orientare il parere scientifico della Commissione Tecnico Scientifica dell’AIFA.
-
Requisiti nazionali da inserire in fase di nazionalizzazione in etichetta esterna di farmaci approvati con procedura centralizzata - Aggiornato al 12/01/2026 [0.6 Mb] [PDF] >
-
Italian Blue-Box requirements to appear in the outer labelling of centralised medicinal products for human use on the italian market - Updated to 12/01/2026 [0.31 Mb] [PDF] >
Uffici di riferimento






